Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2
For Emergency Use Authorization Only | For in vitro diagnostic use | Rx Only
Product Description
On April 24, 2020, the U.S. Food and Drug Administration (FDA) issued an Emergency Use Authorization (EUA) amendment for BGI’s Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2. The amendment expands the previously issued EUA label to further include the use of an automated sample preparation system, additional viral RNA extraction kit, and PCR systems for testing a broader range of clinical samples. Specifically, viral RNA extraction can be processed by the kits manufactured by MGI (a subsidiary of BGI Group) or Qiagen. Also, the highly sensitive SARS-CoV-2 detection test can return results within 4 hours for 192 samples collected from the throat (oropharyngeal) swabs, nasopharyngeal swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasal washes, nasal aspirates, and bronchoalveolar lavage fluid (BALF) using the MGI automated sample preparation system. Hospitals and reference laboratories can run the test on Roche LightCycler 480 Instrument as well as Applied Biosystems 7500 Fast, 7500, and QuantStudio 5 Real-Time PCR Systems.
Features
● Taqman Reverse Transcription PCR
● ORF1ab gene as domain target
● Human β–actin as an internal control
● Manufacturing in ISO 13485 compliant and high-volume production facility
● Stringent QC with positive and no template controls
Benefits
● Highly sensitive – Detect as low as 100 viral copies/mL for BALF samples
● Highly specific – No cross-reactivity with 54 human respiratory pathogens
● High-throughput – Ramp up labs for large-scale, community-based testing
● Fast TAT – Sample to result in 4 hours with automated sample preparation system
● Ease of use – All-inclusive with pre-mixed reaction reagents
● Easy interpretation – Analysis of one target with well-defined controls
Specifications
● 50 reactions per kit
● Acceptable samples collected from the throat (oropharyngeal) swabs, nasopharyngeal swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasal washes, nasal aspirates, and bronchoalveolar lavage fluid (BALF)
● Acceptable real-time PCR systems:
– Applied Biosystems 7500 Fast Real-Time PCR System, Software v2.0.6
– Applied Biosystems 7500 Real-Time PCR System, Software v2.0.5
– Applied Biosystems QuantStudio 5 Real-Time PCR System, 96-Well, Software v1.5.1
– Roche LightCycler 480 Instrument II, 96-Well, Software v1.5.0
● Acceptable viral RNA extraction kits:
– MGIEasy Nucleic Acid Extraction Kit, 96 or 1728 preps
– QIAamp Viral RNA Mini Kit, 50 or 250 preps
● MGISP-960RS Automated Sample Preparation System, Software v1.2 (Optional)
● Limit of detection for BALF samples: 100 viral copies/mL
● Limit of detection for throat swab samples: 150 viral copies/mL
● Reagents stable under dark for 5 days at 2-8°C or 12 months at -18°C
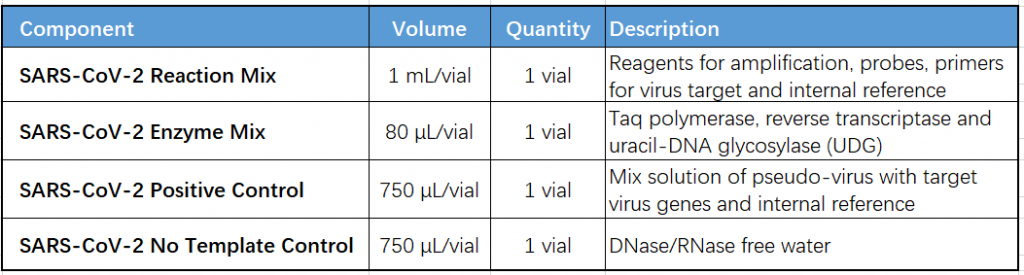
Key Components
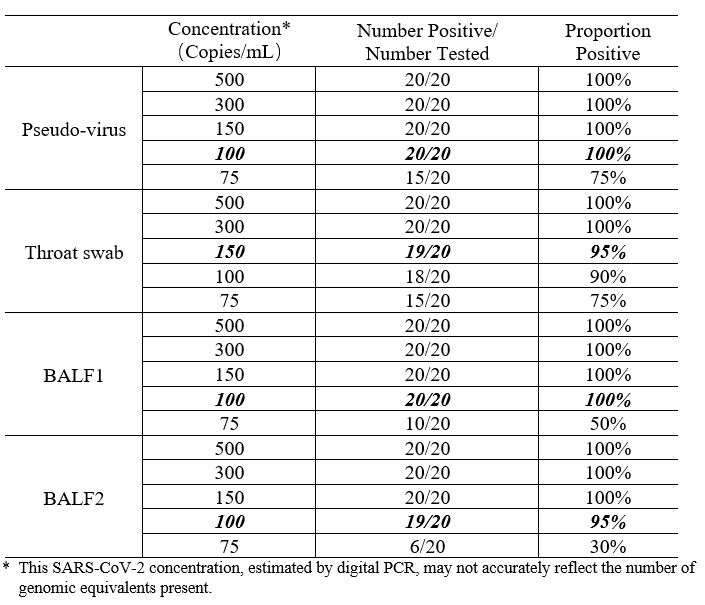
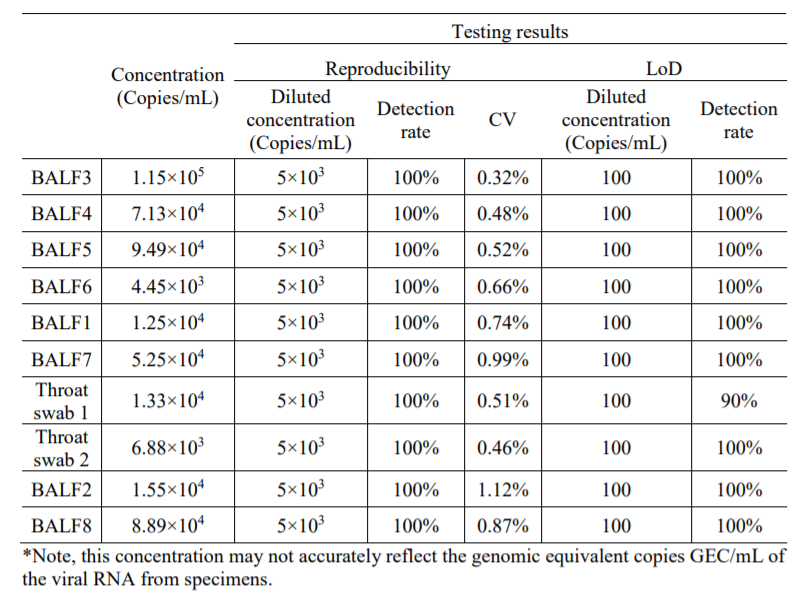
Performance Characteristics
1. Limit of Detection (LoD): 100 copies/mL (BALF); 150 copies/mL (Throat Swabs)
2. Reactivity/Inclusivity
In silico analysis was performed and the assay was mapped to 284 complete SARS-CoV-2 genomes of the human host in GenBank and GISAID databases as of March 10th, 2020. Primer and probes sequences for Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2 had 100% homology to all SARS-CoV-2 isolates analyzed.
3. Analytical Specificity
Cross-reactivity of the Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2 was evaluated using both in silico analysis and by wet testing pathogens. No cross-activity was found in the tested pathogens as shown in the table below.
Intended Use and Emergency Use Authorization Information
The Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2 is an in vitro diagnostic real-time reverse transcription-PCR assay for the qualitative detection of SARS-CoV-2 nucleic acids in throat (oropharyngeal) swabs, nasopharyngeal swabs, anterior nasal swabs, mid-turbinate nasal swabs, nasal washes, nasal aspirates, and bronchoalveolar lavage fluid (BALF) from individuals who are suspected of COVID-19 by their healthcare provider. Testing is limited to laboratories certified under the Clinical Laboratory Improvement Amendments of 1988 (CLIA), 42 USC §263a, to perform high complexity tests.
Test results are for the identification of SARS-CoV-2 RNA. The SARS-CoV-2 RNA is generally detectable in respiratory specimens during the acute phase of infection. Positive results are indicative of the presence of SARS-CoV-2 RNA; clinical correlation with patient history and other diagnostic information is necessary to determine patient infection status. Positive results do not rule out bacterial infection or co-infection with other viruses. The agent detected may not be the definite cause of the disease. Laboratories within the United States and its territories are required to report all positive results to the appropriate public health authorities.
Negative results do not preclude SARS-CoV-2 infection and should not be used as the sole basis for patient management decisions. Negative results must be combined with clinical observations, patient history, and epidemiological information.
The Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2 is intended for use by trained clinical laboratory personnel specifically instructed and trained in the techniques of real-time PCR and in vitro diagnostic procedures. The Real-Time Fluorescent RT-PCR Kit for Detecting SARS-CoV-2 is only for use under the Food and Drug Administration’s Emergency Use Authorization.
COVID-19 Inquiries
Products from BGI that are related to COVID-19 are regulated and only available to clinical and diagnostic laboratories. If you are a consumer looking for a COVID-19 test, please see your health care provider for medical advice. For additional COVID-19 information, visit the CDC or WHO sites.
Related Information
* Check out the Instructions for Use for more details about this product.
* Read the flyer for our product highlights.
* Download our Letter-of-Authorization from FDA.
* Download our healthcare provider fact sheet and patient fact sheet.
* Instructions for Use (English) or Instructions for Use (Francaise) in Canada only.
* Download the Frequently Asked Questions
Contact Us
* Request a quote and your BGI or MGI representative will reply to you within 24 hours.
In the United States:
– This test has not been FDA cleared or approved;
– This test has been authorized by FDA under a EUA for use by authorized laboratories;
– This test has been authorized only for the detection of nucleic acid from SARS-CoV-2, not for any other viruses or pathogens; and
– This test is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of in vitro diagnostic tests for detection and/or diagnosis of COVID-19 under section 564(b)(1) of the Act, 21 U.S.C. § 360bbb- 3(b)(1) unless the authorization is terminated or revoked sooner.