Introducing GPMeta: Ultrarapid GPU-accelerated Pathogen Identification Approach | BGI Insight
2023-04-24
Metagenomic sequencing (mNGS) is a powerful diagnostic tool to detect causative pathogens in clinical microbiological testing. Rapid and accurate classification of metagenomic sequences is a critical procedure for pathogen identification in the dry-lab step of mNGS test. However, this critical step is hampered by classifying sequences within a clinically relevant timeframe.
To address this challenge, a BGI Genomics team led by Xuebin Wang has recently launched GPMeta, an ultra-fast pathogen detection approach, and published these highlights in Briefings in Bioinformatics.
GPMeta can quickly and accurately identify pathogens through complex and massive mNGS sequencing data. Using simulated datasets and metagenomic sequencing datasets from real clinical samples, results were benchmarked against state-of-the-art tools used by the bioinformatics research community such as Bowtie2, Bwa, Kraken2, and Centrifuge.
Results show that GPMeta not only has higher accuracy but also exhibits significantly faster speed. In addition, GPMeta offers a GPMetaC clustering algorithm, a statistical model for clustering and rescoring ambiguous alignments to improve the discrimination of highly homologous sequences from microbial genomes with average nucleotide identity >95%. These results underline GPMeta's key role in the development of the mNGS test in infectious diseases that require rapid turnaround times.
Background
The faster and earlier detection of causative pathogens is critical for precise antibiotic therapy instead of empiric treatment. It can simultaneously detect almost all new and known pathogenic microorganisms in the patient's body in one test and has huge potential applications in infection.
mNGS detection comprises two components: wet-lab experimental manipulations involving clinical sample preprocessing, total nucleic acid extraction, library preparation and sequencing, and dry-lab bioinformatics analysis which includes raw sequencing data preprocessing, removal of human host sequences, sequence alignment to the curated pathogen database and taxonomic classification of microbial sequences.
Bioinformatics analysis is the final crucial step in mNGS detection, which needs to be completed quickly and accurately to accelerate the entire detection process. However, there is an urgent need for new strategies to accelerate the bioinformatics analysis of pathogen identification.
To meet this challenge, GPMeta uses a succinct hash index scheme and supports multiple GPUs to carry out on split databases simultaneously, which meets a growing need for the ability to deal with a rapidly expanding number of microbial genomes.
Method design highlights
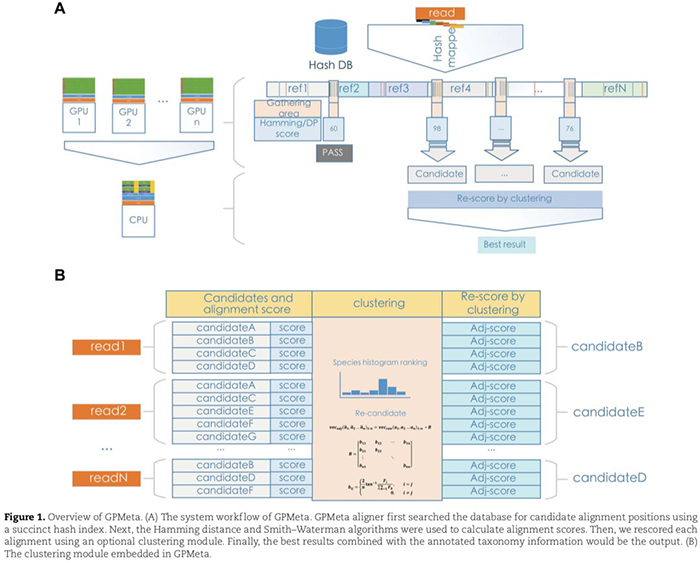
In terms of the alignment process: Firstly, the sequenced reads are sliced into different k-mers, and a hash index algorithm is used to query the hashed pathogen database Hash DB to obtain the corresponding positions of each k-mer. Then, a DetectChain algorithm is used to associate k-mers to determine candidate alignment positions.
The highest corrected score is selected as the optimal sequence alignment position. Finally, an optional clustering model was used to optimize and re-score the alignment scores. In addition, GPMeta allowed for multi-GPU support and the whole alignment process could be distributed to multiple GPUs to carry out.
GPMetaC, as an optional module of GPMeta, adjusted the alignment scores by clustering within batches to improve the discrimination of highly homologous sequences.
Key results
GPMeta and GPMetaC can accurately remove human sequences and detect pathogenic sequences.
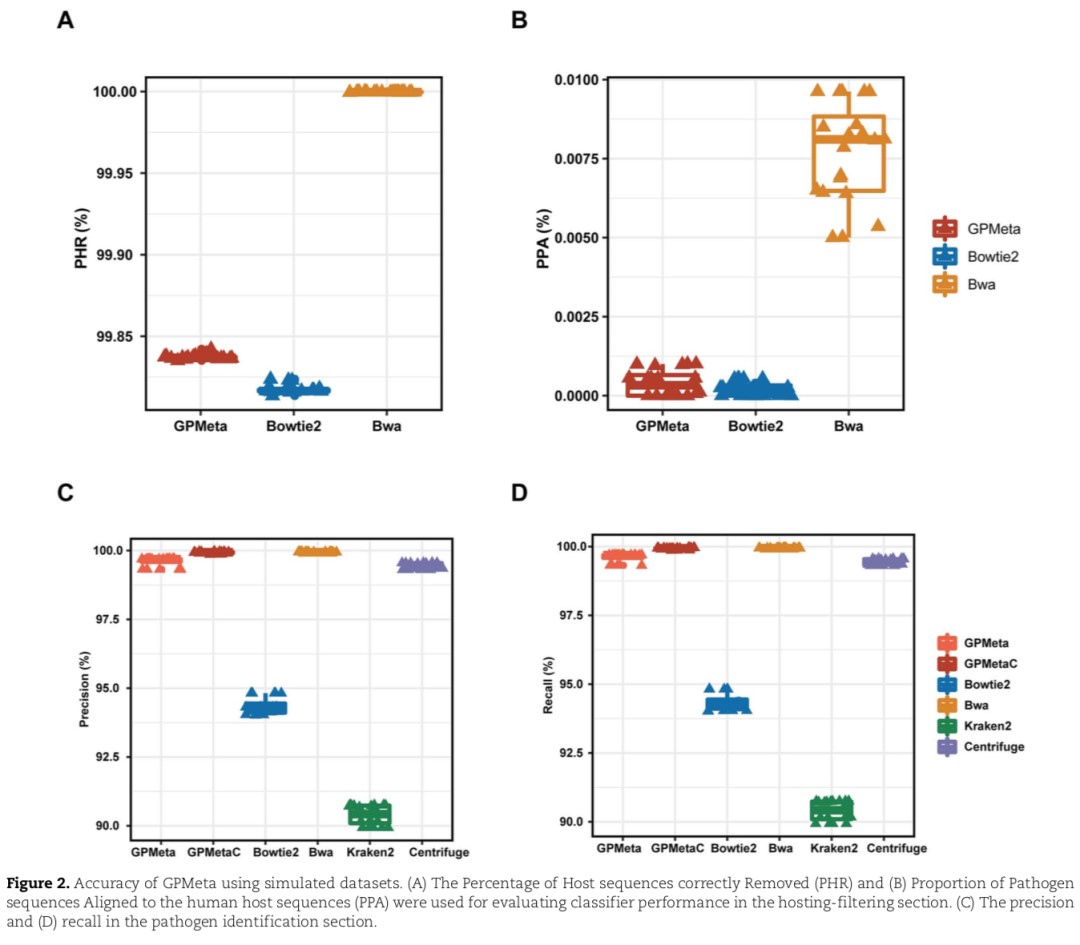
On the accuracy of removing human source sequences (as shown in Figures A and B below):
- Compared with GPMeta and Bowtie2, Bwa has the highest PHR, but it also introduces higher PPA, which means that more pathogenic sequences are mistakenly removed.
- GPMeta and Bowtie2 have similar PHR and PPA, superior to Bwa.
On the accuracy of pathogen detection (Figures C and D below):
- GPMetaC and Bwa have the highest accuracy and recall
- GPMetaC is slightly higher than GPMeta, indicating the impact of GPMetaC on the clustering correction function.
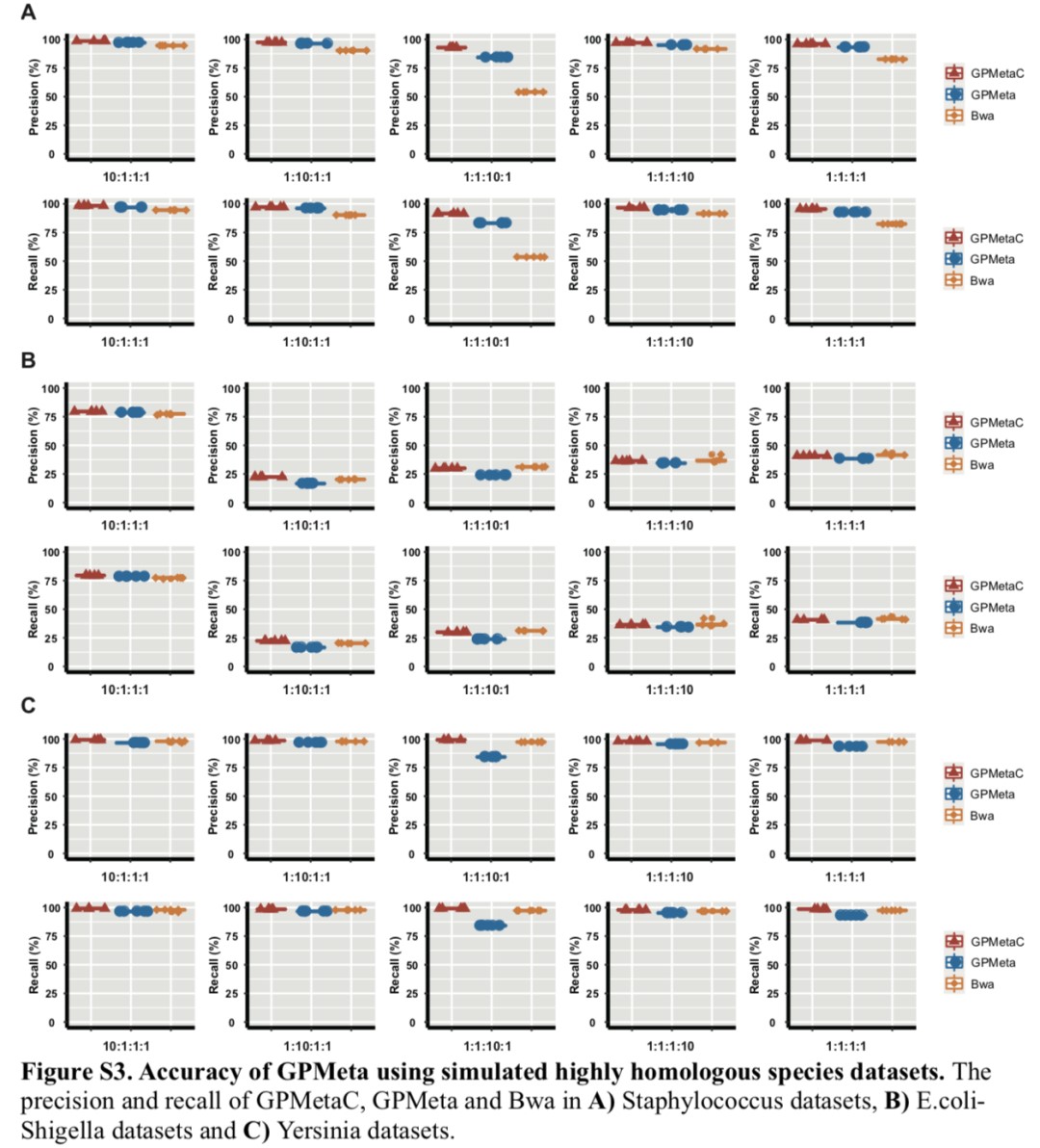
GPMetaC can accurately detect and distinguish highly homologous species:
► Select three genera with a similarity of 85-97%, Staphylococcus (Figure A below)
Escherichia coli (Figure B below) and Yersinia (Figure C below) as the simulation data set
The accuracy of GPMetaC is better than that of Bwa in Figures A and C, while the performance of both is consistent in Figure B.
In terms of accuracy and recall, GPMetaC has improved by about 2% compared to GPMeta, demonstrating the effective performance of the clustering correction function in correcting ambiguity alignment sequences.
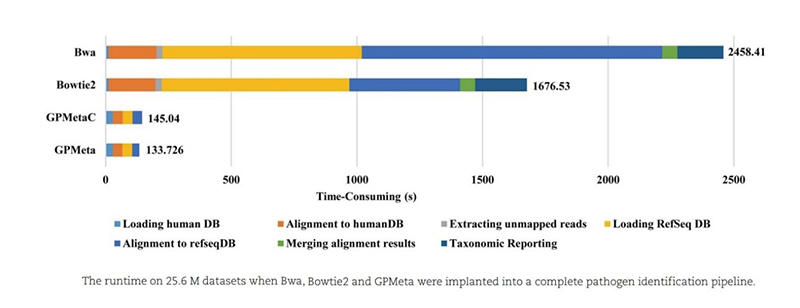
Comparison of software running speed:
On the 25M reads dataset, GPMeta and GPMetaC only need less than 3 minutes to complete the entire detection analysis.
On the 110M reads dataset (conventional mNGS detection data volume), GPMeta and GPMetaC only need 4 minutes to complete the entire detection analysis.
Compared to the entire 190Gb pathogen library, GPMeta and GPMetaC accelerated by 39-50 and 12-35 times, respectively, compared to Bwa and Bowtie2.
The entire detection and analysis GPMeta are 18 times and 12 times faster than Bwa and Bowtie2, respectively.
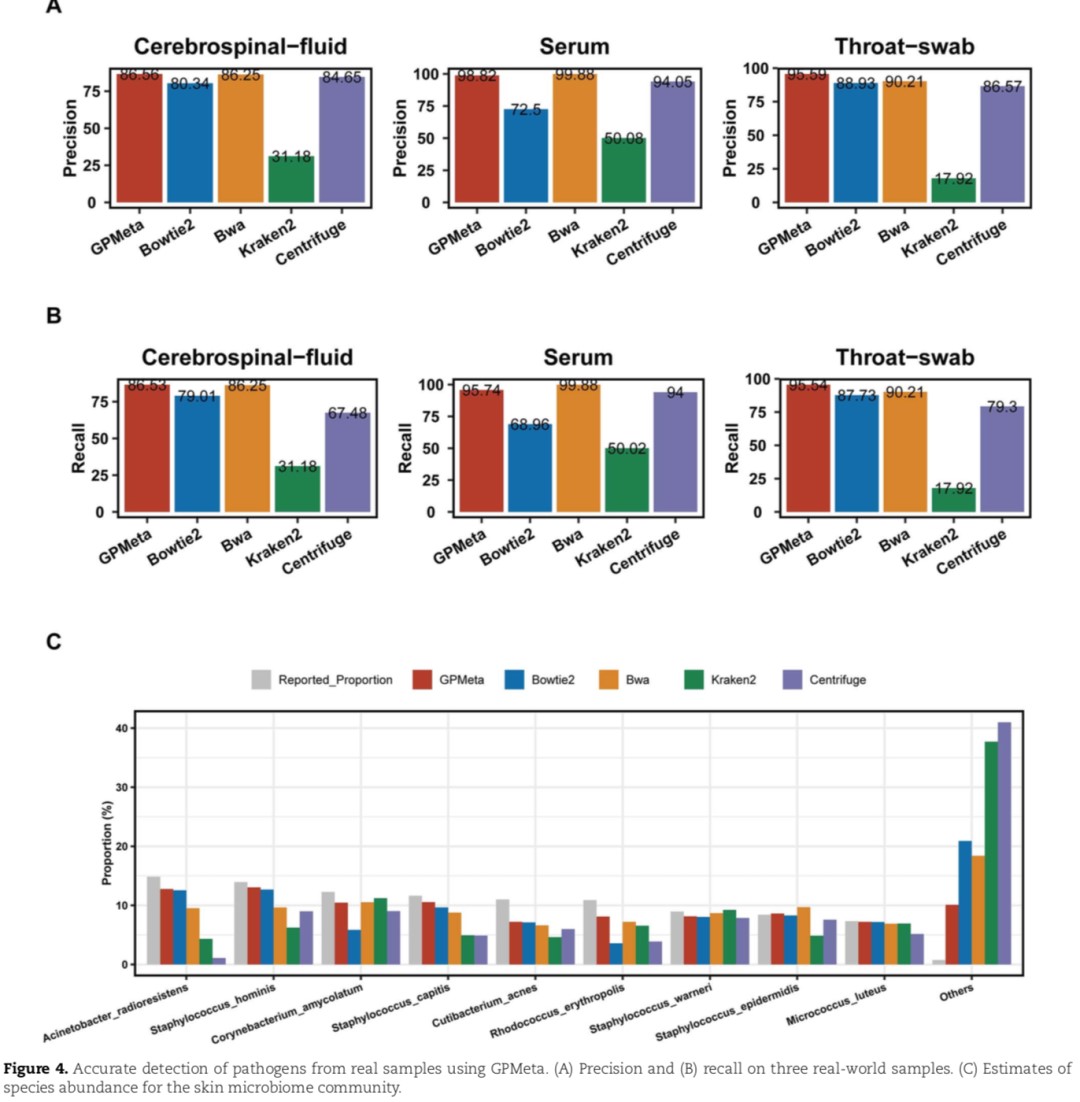
GPMeta has the highest accuracy for clinical samples:
Dataset evaluation of three types of tissue samples - cerebrospinal fluid, blood, and nasopharyngeal swabs - was separately conducted.
The standard for determining the true microbial sequence in the evaluation is: BLASTn compares it to the pathogen sequence library, and sequences that meet the E-value cutoff of 1x10-20, identity>95%, and match rate>0.8 are determined to come from the true microbial species. The results show that GPMeta and Bwa display the highest accuracy and recall, outperforming other commonly used software (Figures A and B below).
► In the skin microbial sample data set, GPMeta shows the best species richness estimation, and its abundance results are closer to the species richness reported in the original literature (Figure C below).
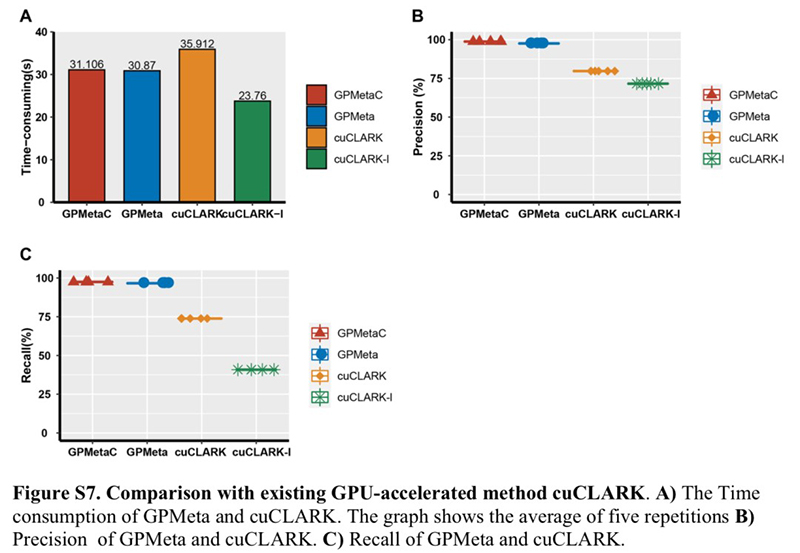
GPMeta is superior to the existing GPU-accelerated microbial detection tool cuCLARK:
Figure A shows that on a 20M reads simulated dataset, both GPMeta and cuCLARK full mode can complete pathogen library comparison in half a minute, while cuCLARK light mode is faster. In terms of accuracy, as shown in Figures A and B, GPMeta/GPMetaC can achieve an accuracy and recall rate of 96 to 98%. However, the accuracy and recall rate of both cuCLARK operating modes is less than 80%.
Conclusion
GPMeta is a powerful tool to timely and accurately identify pathogens from mNGS data, which is of great importance in eliminating the threat from severe acute infections and in targeting precise and effective antibiotic therapy.
Moreover, GPMeta supports multiple GPUs to perform alignment and taxonomic classification of microbial sequences on split databases simultaneously and automatically merges results from multiple sub-databases, which is significant to keep up with the rapidly expanding microbial genome database. To make the best use of GPMeta, how to best and easily integrate it into clinical practices needs further study.
We welcome non-commercial users to test-drive GPMeta at https://github.com/Bgi-LUSH/GPMeta.
About BGI Genomics
BGI Genomics, headquartered in Shenzhen China, is the world's leading integrated solutions provider of precision medicine. Our services cover over 100 countries and regions, involving more than 2,300 medical institutions. In July 2017, as a subsidiary of BGI Group, BGI Genomics (300676.SZ) was officially listed on the Shenzhen Stock Exchange.